
1)About intracranial aneurysm
Intracranial aneurysm (IA) is a lesion characterized by the regional bulging of intracranial arteries mainly at their bifurcation sites and by degenerative changes of arterial walls with the inflammatory infiltrates. IA affects 3 to 5 % of general public and is the primary cause of subarachnoid hemorrhage, which is the most severe form of stroke with the mortality rate of 50 %. Given such a poor prognosis of subarachnoid hemorrhage once after the onset, prevention of formation, growth and rupture of IA is socially important. However, the current option of treatment interventions targeting IAs is limited only to surgical ones like microsurgical clipping or endovascular coiling because no drug therapy is currently available for IAs, leaving many patients without treatment. Therefore, elucidation of underlying mechanisms of IA and development of therapy targeted to its pathogenesis are urgently needed.
2)Findings from our research works
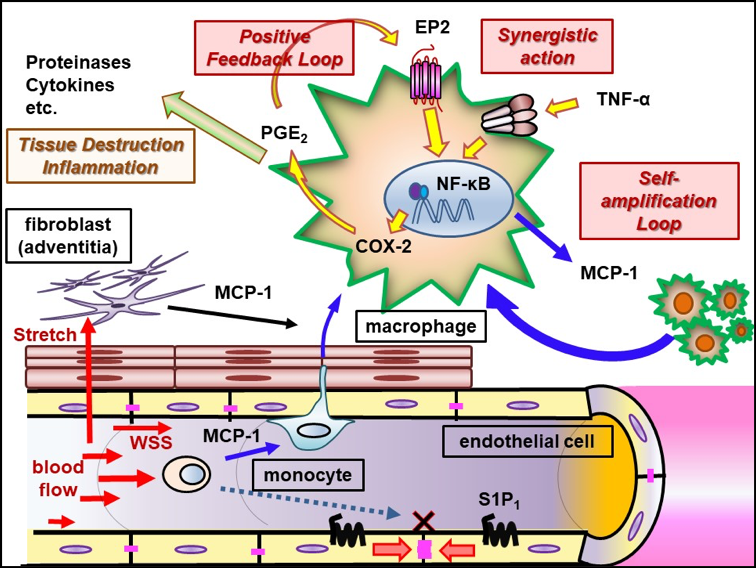
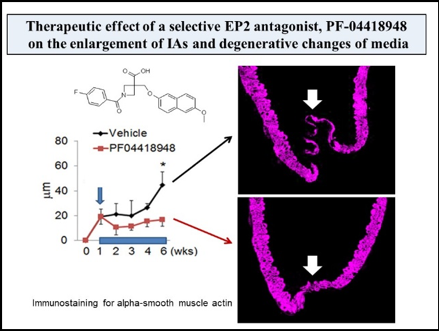
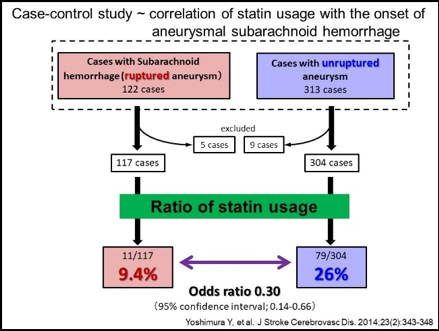
Mainly using a rodent model of IAs, we have clarified some important machineries underlying the formation and progression of the disease. In brief, high wall shear stress loaded at the prospective site of IA formation stimulates endothelial cells to activate a transcription factor, NF-kB. The activation of NF-B promotes the production of MCP-1 from endothelial cells. Monocytes in blood flow are then recruited into arterial walls via MCP-1. In this step, another mechanical force from blood flow, mechanical stretch, activates fibroblasts present at adventitia of arterial walls especially at their bifurcation sites to produce MCP-1. MCP-1 from endothelial cells and fibroblasts cooperatively recruits monocyte into the adventitia of intracranial arterial walls. Macrophages differentiated from monocyte produce various pro-inflammatory factors like cytokines and proteinases to evoke inflammation and induce tissue damage. In this process, some specific mechanisms function in macrophages to make inflammation being long-lasting, amplified and become chronic. TNF-alpha or other cytokines present in the microenvironment of IA lesions activates NF-kB in macrophages and NF-kB activation in turn induces COX-2 expression. COX-2 induced produces prostaglandin E2 and prostaglandin E2 then acts on its receptor subtype, EP2, to activate NF-kB. Thereby, the positive feedback loop including prostaglandin E2-EP2- NF-kB-COX-2 is formed, making inflammatory responses once triggered being long-lasting and exacerbated. Also, EP2 signaling synergizes with TNF-alpha to amplify expression of pro-inflammatory genes induced by this cytokine via forming transcription factor complex or induction of RNA-binding protein, HuR. Furthermore, MCP-1 production from macrophages under NF-kB and HuR activation forms the self-amplification loop among macrophages. Here, because NF-kB, EP2 or some other molecules involved in inflammatory responses in the microenvironment of the disease mediates the chronic inflammation leading to the progression of the disease, these molecules can be therapeutic targets to treat IAs to prevent progression or rupture. Indeed, drugs targeting these molecules can prevent the formation or the progression of IAs in rodent models. For example, PF-04418948, a selective EP2 antagonist, can suppress further enlargement of the pre-existing IAs induced in a rat model. Also, stains with a potent inhibitory effect against NF-kB activation can suppress the progression of the disease in a rat model. Furthermore, the usage of statins negatively correlates with the onset of subarachnoid hemorrhage due to rupture of IAs in human cases, suggesting the inhibitory effect of statins on rupture of IAs.
3)Current research topics
① Mechanisms regulating rupture of IAs
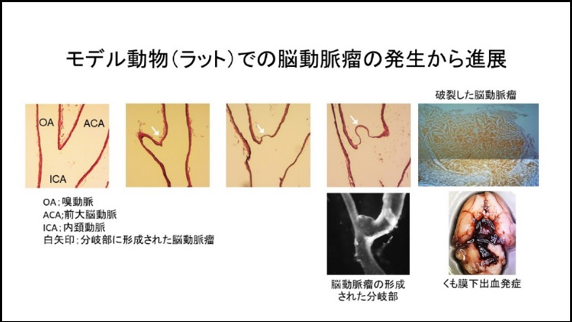
Recently, we have established a rat model of IAs in which more than half of IAs induced spontaneously rupture during the observation period resulting in the onset of subarachnoid hemorrhage. The pathological findings of IAs or clinical course after the onset of subarachnoid hemorrhage well mimic human ones. We can thus examine machineries underlying rupture of IAs using this newly-established model. Most recently, we have clarified the formation of vasa vasorum at the adventitia of IAs and the accumulation of inflammatory cells including macrophages and neutrophils around this structure as events related with rupture of the lesions.
We have been studying molecular machineries regulating rupture of IAs and also been examining effect of some drugs on rupture of lesions.
② Establishment of ‘macrophage imaging’ techniques
IAs are usually founded without any neurological symptoms in brain dock or out-patient’s clinics using magnetic resonance imaging. Indication of surgical intervention to such an incidentally-found unruptured IAs should be carefully determined considering confounding factors such as patient’s background, nature of the lesion and risk of surgical intervention because most IAs are asymptomatic lesions. Among several factors to be considered, morphological parameters such as size, location and shape of the lesion are of clinical importance for risk assessment of rupture and of surgical manipulations. Although most of IAs is considered as a silent lesion with less inflammatory or degenerative changes in lesions, some of them follow rather unstable course including the enlargement or rupture. However, until recently, there is not any accurate method available to predict rupture risk. Based on above background, we have been developing a novel nano-particle based contrast agent for detecting macrophages in MRI.
③ Molecular mechanisms regulating hemodynamic force-sensing
IA is a hemodynamic force-regulated disease. High wall shear stress loaded at bifurcation sites of intracranial arteries where IAs are formed triggers the formation of the disease and excessive low wall shear stress and concomitant turbulent flow presumably regulate event leading to the progression of the disease. We have found that these hemodynamic forces at least partially activate endothelial cells in situ to trigger inflammatory responses. However, many remain to be elucidated; e.g. how hemodynamic force activates endothelial cells or what is a critical factor regulating hemodynamic force-mediated induction of inflammatory responses.We have thus examined molecular machineries how hemodynamic force regulates the pathogenesis of various disease by using an IA as a model of such a hemodynamic force-mediated disease.
Selected review articles
1. Aoki T#, Miyata H, Abekura Y, Koseki H, Shimizu K. Rat model of intracranial aneurysm. Variations, usefulness and limitations of the Hashimoto Model. Acta Neurochir. Suppl. 2020;127:35-41. # corresponding author
2. Kushamae M, Shimizu K, Aoki T#. Macrophage Imaging of Intracranial Aneurysms. Neurol. Med. Chir. (Tokyo) 2019;59(7):257-263. # corresponding author
3. Shimizu K, Kushamae M, Mizutani T, Aoki T#. Intracranial Aneurysm as a Macrophage-Mediated Inflammatory Disease. Neurol. Med. Chir. (Tokyo) 2019;59(4):126-132. # corresponding author
4. Aoki T, Yamamoto R, Narumiya S. Targeting macrophages to treat intracranial aneurysm. Oncotarget 2017;8(62):104704-104705.
5. Aoki T, Nozaki K. Preemptive medicine for cerebral aneurysm. Neurol. Med. Chir. (Tokyo) 2016;56(9):552-568.
6. Fukuda M, Aoki T#. Molecular basis for the intracranial aneurysm formation. Acta Neurochir. Suppl. 2015;120:13-15. # corresponding author
7. Aoki T#, Fukuda M, Narumiya S. Chronic inflammation in intracranial aneurysm formation. Inf. Reg. 2013;33(5):283-287. # corresponding author
8. Aoki T, Narumiya S. Prostaglandins and chronic inflammation. Trends Pharmacol. Sci. 2012;33(6):304-311.
Selected references
1. Koseki H, Miyata H, Shimo S, Ohno N, Mifune K, Shimano K, Yamamoto K, Nozaki K, Kasuya H, Narumiya S, Aoki T#. Two diverse hemodynamic forces, a mechanical stretch and a high wall shear stress, determine intracranial aneurysm formation. Transl. Stroke Res. in press # corresponding author
2. Shimizu K, Miyata H, Abekura Y, Oka M, Kushamae M, Kawamata T, Mizutani T, Kataoka H, Nozaki K, Miyamoto S, Aoki T#. High fat diet intake promotes the enlargement and degenerative changes in the media of intracranial aneurysms in rats. J. Neuropathol. Exp. Neurol. 2019;78(9):798-807. # corresponding author
3. Miyata H, Imai H, Koseki H, Shimizu K, Abekura Y, Oka M, Kawamata T, Matsuda T, Nozaki K, Narumiya S, Aoki T#. Vasa vasorum formation is associated with rupture of intracranial aneurysms. J. Neurosurg. 2019;16:1-11. # corresponding author
4. Aoki T#, Koseki H, Miyata H, Itoh M, Kawaji H, Takizawa K, Kawashima A, Ujiie H, Higa T, Minamimura K, Kimura T, Kasuya H, Nozaki K, Morita A, Sano H, Narumiya S. RNA sequencing analysis revealed the induction of CCL3 expression in human intracranial aneurysms. Sci. Rep. 2019;9(1):10387. # corresponding author
5. Miyata H, Shimizu K, Koseki H, AbekuraY, Kataoka H, Miyamoto S, Nozaki K, Narumiya S, Aoki T#. Real-Time Imaging of an Experimental Intracranial Aneurysm in Rats. Neurol. Med. Chirur. (Tokyo) 2019;59(1):19-26. # corresponding author
6. Aoki T, Saito M, Koseki H, Tsuji K, Tsuji A, Murata K, Kasuya H, Morita A, Narumiya S, Nozaki K; for MR macrophage imaging study investigators. Macrophage imaging of cerebral aneurysms with ferumoxytol: an exploratory study in an animal model and in patients. J. Stroke Cerebrovasc. Dis. 2017;26(10):2055-2064.
7. Aoki T, Frosen J, Fukuda M, Bando K, Shioi G, Tsuji K, Ollikainen E, Nozaki K, Laakkonen J, Narumiya S. Prostaglandin E2-EP2-NF-kB signaling in macrophages as a potential therapeutic target of intracranial aneurysm. Sci. Signal. 2017;10:eaah6037.
8. Miyata H, Koseki H, Takizawa K, Kasuya H, Nozaki K, Narumiya S, Aoki T#. T cell function is dispensable for intracranial aneurysm formation and progression. PLoS ONE 2017;12(4):e0175421. # corresponding author
9. Yamamoto R, Aoki T, Koseki H, MFukuda M, Hirose J, Tsuji K, Takizawa K, Nakamura S, Miyata H, Hamakawa N, Kasuya H, Nozaki K, Hirayama Y, Aramori I, Narumiya S. An S1P1 agonist, ASP4058, suppresses intracranial aneurysm progression through promoting endothelial integrity and blocking macrophage transmigration. Br. J. Pharmacol. 2017;174(13):2085-2101.
10. Aoki T#, Yamamoto K, Fukuda M, Shimogonya Y, Fukuda S, Narumiya S. Sustained expression of MCP-1 by low wall shear stress loading concomitant with turbulent flow on endothelial cells of intracranial aneurysm. Acta Neuropathol. Commun. 2016;4(1):48. # corresponding author
11. Aoki T#, Fukuda M, Nishimura M, Nozaki K, Narumiya S. Critical role of TNF-alpha-TNFR1 signaling in intracranial aneurysm formation. Acta Neuropathol. Commun. 2014;2:34. # corresponding author
12. Fukuda M, Aoki T, Manabe T, Maekawa A, Shirakawa T, Kataoka H, Takagi Y, Miyamoto S, Narumiya S. Exacerbation of intracranial aneurysm and aortic dissection in hypertensive rat by treatment with a prostaglandin F receptor antagonist, AS604872. J. Pharmacol. Sci. 2014;126(3):230-242.
13. Yoshimura Y, Murakami Y, Saitoh M, Yokoi T, Aoki T, Miura K, Ueshima H, Nozaki K; SSS Research Group Statin Use and Risk of Cerebral Aneurysm Rupture. A Hospital-Based Case-Control Study in Japan. J. Stroke Cerebrovasc. Dis. 2013;23(2):343-348.
14. Aoki T, Kataoka H, Nishimura M, Ishibashi R, Morishita R, Miyamoto S. Regression of intracranial aneurysms by simultaneous inhibition of NF-kB and ets with chimeric decoy oligodeoxynucleotide treatment. Neurosurgery 2012;70(6):1534-1543.
15. Aoki T, Nishimura M, Matsuoka T, Yamamoto K, Furuyashiki T, Kataoka H, Kitaoka S, Ishibashi R, Ishibazawa A, Miyamoto S, Morishita R, Ando J, Hashimoto N, Nozaki K, Narumiya S. PGE2-EP2 signaling in endothelium is activated to hemodynamic stress and induces cerebral aneurysm through an amplifying loop via NF-B. Br. J. Pharmacol. 2011;163(6):1237-1249.
16. Aoki T, Nishimura M, Kataoka H, Ishibashi R, Nozaki K, Miyamoto S. Complementary inhibition of cerebral aneurysm formation by eNOS and nNOS. Lab. Invest. 2011;91(4):619-626.
17. Aoki T, Nishimura M, Kataoka H, Ishibashi R, Takagi Y, Hashimoto N. Toll like receptor 4 expression during cerebral aneurysm formation. J. Neurosurg. 2010;113(8):851-858.
18. Aoki T, Nishimura M, Kataoka H, Ishibashi R, Nozaki K, Hashimoto N. Reactive oxygen species are responsible for cerebral aneurysm formation: study using p47phox-/- mice and the free radical scavenger ‘edaravone’. Lab. Invest. 2009;89(7):730-741.
19. Aoki T, Kataoka H, Ishibashi R, Nozaki K, Morishita R, Hashimoto N. Reduced collagen biosynthesis in cerebral aneurysmal walls is mediated by up-regulated expression of interleukin-1β and the nuclear factor-κB pathway. Arterioscler. Thromb. Vasc. Biol. 2009;29(7):1080-1086.
20. Aoki T, Kataoka H, Ishibashi R, Nakagami H, Nozaki K, Morishita R, Hashimoto N. Pitavastatin suppresses the formation and progression of cerebral aneurysms through the inhibition of nuclear factor kappa B pathway. Neurosurgery 2009;64(2):357-366.
21. Aoki T, Kataoka H, Ishibashi R, Nozaki K, Hashimoto N. Impact of monocyte chemoattractant protein-1 deficiency on cerebral aneurysm formation. Stroke 2009;40(3):942-951.
22. Aoki T, Kataoka H, Ishibashi R, Nozaki K, Hashimoto N. Cathepsin B, K and S are expressed in cerebral aneurysms and promote the progression of cerebral aneurysms Stroke 2008;39(9):2603-2610.
23. Aoki T, Kataoka H, Ishibashi R, Nozaki K, Hashimoto N. Simvastatin suppresses the progression of experimentally induced cerebral aneurysms in rats. Stroke 2008;39(4):1276-1285.
24. Aoki T, Kataoka H, Shimamura M, Nakagami H, Wakayama K, Moriwaki T, Ishibashi R, Nozaki K, Morishita R, Hashimoto N. NF-B is a key mediator of cerebral aneurysm formation. Circulation 2007;116(24):2830-2840.
25. Aoki T, Kataoka H, Moriwaki T, Nozaki K, Hashimoto N. The role of TIMP-1 and TIMP-2 in the progression of cerebral aneurysms. Stroke 2007;38(8):2337-2345.
26. Aoki T, Kataoka H, Morimoto M, Nozaki K, Hashimoto N. Macrophage-derived matrix metalloproteinase-2 and -9 promote the progression of cerebral aneurysms in rats. Stroke 2007;38(1):162-169.