
ミトコンドリアでのエネルギー産生機構の機能不全が根本原因のミトコンドリア心筋症、ミトコンドリア病は有効な治療法がない難病ですが、病態の進行増悪に至る過程はこれまで十分には解明されていませんでした。
我々は国循で診療されたミトコンドリア心筋症の患者組織を用いた遺伝子発現解析とミトコンドリア心筋症モデルマウスの心臓を用いた単一細胞遺伝子発現解析を組み合わせ、ミトコンドリア心筋症の進行過程を擬似的にトレースすることに成功しました。病初期にはミトコンドリアの機能低下を補填するため、ミトコンドリアの数を増やす代償機構が機能しますが、Atf3はその機構をスイッチオフさせ、病態を進行させることを明らかにしました。
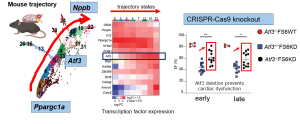
病態進行因子を明らかにするため、二つのアプローチを用いました。まず国循で診療された8ヶ月齢女児のミトコンドリア心筋症患者組織を用いた遺伝子発現解析を実施しました。超重症心不全で補助循環が必要となる状態でしたが、予想外に心筋組織は不均一で、変性の少ない心筋(図:組織像の左半分)と変性が多く進行した心筋(図:組織像の右半分)が混在していました。組織切片上で遺伝子発現解析を実施したところ、心不全で上昇する遺伝子Nppbは、組織切片の右側に強く、左側では上昇しておらず、組織切片所見に合致するものでした。これは比較的変性の少ない左側から右側への進行を想像させるものであり、左側で強く発現する遺伝子を探索したところ、Atf3を見出しました。患者組織を調整し、単一細胞レベルの遺伝子発現解析も進めたところ、Atf3の発現はミトコンドリアの数を増やすように働くPpargc1aの発現とともに低下していき、Nppbを発現する不全心筋に移行する様子を思わせる病態進行過程が導き出されました(図右)。

ヒトミトコンドリア心筋症患者で見出した知見を検証するため、ミトコンドリア心筋症モデルマウス(FS6KDマウス)の心臓を用いて単一細胞遺伝子発現解析を実施しました。モデルマウスでは複数のタイミングで検討を進めたところ、心機能低下がマイルドな生後4−8週齢の病気早期のマウス心臓には、ミトコンドリアを増やすPpargc1aを非常に強く発現している心筋細胞が多く、代償期と考えられました。しかしその中に少数ですが、すでに不全心筋のマーカーであるNppbの発現が認められる細胞群が混じっており、不均一性が高いことがわかりました。モデルマウスの1細胞遺伝子発現解析では、細胞数も多く解像度の高い解析が可能であったため、擬似的に導き出した病態進行過程を描出し、細胞の運命決定に関わる転写因子に注目して解析したところ、病気早期と進行期を結ぶ移行期に発現上昇する遺伝子としてAtf3を同定しました。
ヒト組織でも同様にAtf3が注目分子として上がってきたことより、Atf3を先天的に欠損するマウスを作成しました(図右、Atf3-/-FS6KD)。このマウスでは、病態進行の抑制により、心機能低下の抑制、すなわち心保護効果が見られました。これらの成果から、難病であるミトコンドリア心筋症、ミトコンドリア病の発症メカニズムの理解を深め、今後の治療薬開発につながることも期待されます(Science Advances 2025.)。https://www.science.org/doi/10.1126/sciadv.adq1575
今回採用した研究手法は、ミトコンドリア心筋症のみならず、他の心血管疾患に適用可能です。興味を持ってくれる方の参加をお待ちしています。